





24小時服務熱線:
400-1846-454


第一部分:激光產品FDA檢測
21CFR 1040.10的全稱為《聯邦法規》第21章,第1040.10條款激光產品要求。其本質類似于IEC 60825-1和GB 7247.1,是激光產品安全等級的測試和判定方法,以及激光產品防護要求和標簽說明要求的標準。但在美國其以法規的形式作為要求,做分級上來說比標準的等級要更高。
FDA作為美國輻射放射產品的管制機構,其對激光輻射產品的要求及遵循 21CFR 1040.10
I類激光產品 指激光產品在操作過程中,任何時候人類接觸激光輻射水平不超過21 CFR Subchapter J Part 1040.10表I中規定發射限制。I類級別的激光輻射不被認為是危險的。
II類激光產品
指激光產品在操作過程中,任何時候人類接觸的可見激光輻射水平超過了21 CFR Subchapter J Part 1040.10表II-A中包含輻射限制,但未超過 21 CFR Subchapter J Part 1040.10的表II中包含的激光輻射水平。II 級激光輻射被認為是一種具有慢性觀看危害激光。
IIIa類激光產品
指激光產品在操作過程中,任何時候人類接觸的可見激光輻射水平超過了21 CFR Subchapter J Part 1040.10表II中包含的輻射限制,但未超過21 CFR Subchapter J Part 1040.10表III-A中的輻射發射限制。根據輻照度,IIIa光激光輻射被認為是急性光束內觀察危險或慢性觀察危險,如果直接用光學儀器觀察,則認為是急性觀察危險。
IIIb類激光產品
指激光產品在操作過程中,任何時候人類接觸的可見激光輻射水平超過了21 CFR Subchapter J Part 1040.10表III-A中包含的輻射限制,但未超過21 CFR Subchapter J Part 1040.10表III-B中的輻射發射限制。IIIb級激光輻射被認為是直接輻射對皮膚和眼睛的急性危害。IIIb類激光產品可能具有可拆卸面板,當這些面板移位時,可接觸激光等級可能處于II類到IV類的激光輻射水平。
IV 類激光產品
指激光產品在操作過程中,任何時候人類接觸的可見激光輻射水平超過了21 CFR Subchapter J Part 1040.10表III-B中包含的輻射限制。IV 級激光輻射被認為是直接輻射和伴隨輻射對皮膚和眼睛的急性危害。IV 類激光產品可能具有可拆卸面板,當這些面板移位時,可接觸激光等級可能處于II類到IV類的激光輻射水平。
FDA與2007年發布了Laser Notice No. 50公告,以下21 CRF 1040.10部分章節的要求可以采用EIC 60825-1和IEC 60601-2-22的等同要求替代:
1040.10(b) Definitions
1040.10(c)(1) Classification
1040.10(d) Accessible emission limits
1040.10(e) Tests for determination of compliance
1040.10(f)(1) Protective housing
1040.10(f)(2) Safety interlocks
1040.10(f)(3) Remote Interlock connector
1040.10(f)(4) Key control
1040.10(f)(5) Laser radiation emission indicator
1040.10(f)(6) Beam attenuator
1040.10(f)(7) Location of controls
1040.10(f)(8) Viewing optics
1040.10(f)(9) Scanning safeguard
1040.10(g) Labeling requirements
1040.10(h)(1) User information
1040.11(a) Medical laser products
但以下內容不得替代,依然需滿足21CRF1040.10:
1010.2 Certification
1010.3 Identification
1010.4 Variances
1040.10(a) Applicability
1040.10(c)(2) Removable laser systems
1040.10(f)(10) Manual reset mechanism
1040.10(h)(2) Purchasing and servicing information
1040.10(i) Modification of a certified product
1040.11(b) Surveying, leveling and alignment laser products
1040.11(c) Demonstration laser products
總結:
直接按照21CFR 1040.10檢測,是目前符合FDA的最直接方式。如果已經按IEC 60825或IEC60601-2-22檢測,則應增加上述不可替代部分的21 CFR1040.10對應條款檢測。
質量保證:實驗室嚴格按照ISO 17025進行質量體系管理,檢測過程控制精準,結果質量保證;
第二部分:激光產品FDA注冊
激光FDA即制造商按照美國法典下的聯邦食品、藥物和化妝品法案下的電子產品輻射控制規定21CFR 1000-1050中的放射衛生規定,需完成激光類產品記錄和報告事項。同時,美國食品藥品監督管理局(FDA):負責管理輻射電子產品,保護公眾免受電子產品輻射的有害和不必要的暴露。激光產品FDA注冊,即向FDA報告激光產品的質量、設計、放射水平、生產記錄等。
上述激光FDA注冊即報告需含如下信息:
(a)產品類別屬性。
(b)產品信息(名稱、型號、標簽位置)。
(c)產品結構、組件、以及影響輻射量的因素。
(d)每種型號的功能,影響輻射的運行特性以及預期用途和已知用途。
(e)每種型號與電子產品輻射安全有關的標準和設計規范。
(f)每種型號描述產品所包含的物理和電氣特性(例如屏蔽或電子電路)。
(g)輻射安全的測試和測量時所采用的方法和程序,包括控制不必要的或泄漏的輻射參量,以及選擇此類測試程序的依據。 以及產品質量控制程序說明。
(h)對于那些隨著時間的推移會產生更多輻射的產品,請描述所使用的控制方法和程序,以及每種型號在電子產品輻射安全方面的耐久性和穩定性的測試頻率。包括選擇此類方法和程序的基礎,或確定不需要此類測試和質量控制程序的基礎。
(i)提供足夠的根據本節(g)和(h)所述的測試、測量和質量控制程序的結果,以使FDA能夠確定這些測試方法和程序的有效性。
(j)每種型號的所有與電子產品輻射安全有關的認證標簽、警告標志、銘牌以及安裝,操作和使用說明。
(k)其他特定產品要求信息。
特別提醒:如上法規規定的激光產品FDA注冊可知,檢測過程描述和檢測結果是注冊最重要的信息之一,也即要提供21CFR 1040.10檢測報告。中為檢驗技術擁有一流激光實驗室,出具檢測報告可以直接用于激光FDA注冊。
產品報告FDA后將的到收信回執(紙質或郵件)。
回執旨在通知報告人:報告已經收到并已錄入數據庫,并分配報告登錄號。
登錄號是FDA數據庫中報告的唯一標識,有助于特定報告的查取。此外,FDA使用登錄號來確認制造商至少已符合產品的自我聲明符合認證和報告的要求。
登錄號表示FDA已收到報告信,不表示FDA已批準和認可了相關產品,也不表示報告內容的合規和完整新。
")
特別提醒:FDA注冊執行先分配注冊號,后隨即抽查的監督措施。也即提交虛假資料也能獲得注冊號,但一旦被查收,注冊號即有被取消的風險,企業也將面臨黑名單風險。中為檢驗技術以嚴謹真實的原則,以符合性檢測為前提,為企業提供合規的激光產品FDA注冊。
第三部分:激光FDA年報
FDA規定每年的年度報告應在每年的 9 月 1 日前完成。該報告應涵蓋從上一年 7 月 1 日到今年 6 月 30 日期間的產量、庫存量和銷往美國的數量。
每年 6 月 30 日至 9 月 1 日之間有 2 個月的“寬限”期。例如,在 2021 年 9 月 1 日之前提交的 2020 年 7 月 1 日至 2021年 6 月 30 日的年度報告在 2022 年 9 月 1 日之前仍然有效。
也即:每年的7月1日至9月1日這兩個月期間,應該把上年度下半年和本年度上半年的情況進行年報。
節點:在7月1日之后做的新注冊那就下年度再年報,之前注冊的,那當年的9月1號前要完成年報。
年報即報告產品質量情況和年度生產銷售記錄,除了首次注冊需要的信息全部需要外,還需增加生產和銷售記錄:
(a)產品類別屬性。
(b)產品信息(名稱、型號、標簽位置)。
(c)產品結構、組件、以及影響輻射量的因素。
(d)每種型號的功能,影響輻射的運行特性以及預期用途和已知用途。
(e)每種型號與電子產品輻射安全有關的標準和設計規范。
(f)每種型號描述產品所包含的物理和電氣特性(例如屏蔽或電子電路)。
(g)輻射安全的測試和測量時所采用的方法和程序,包括控制不必要的或泄漏的輻射參量,以及選擇此類測試程序的依據。 以及產品質量控制程序說明。
(h)對于那些隨著時間的推移會產生更多輻射的產品,請描述所使用的控制方法和程序,以及每種型號在電子產品輻射安全方面的耐久性和穩定性的測試頻率。包括選擇此類方法和程序的基礎,或確定不需要此類測試和質量控制程序的基礎。
(i)提供足夠的根據本節(g)和(h)所述的測試、測量和質量控制程序的結果,以使FDA能夠確定這些測試方法和程序的有效性。
(j)每種型號的所有與電子產品輻射安全有關的認證標簽、警告標志、銘牌以及安裝,操作和使用說明。
(k)其他特定產品要求信息。
另外增加:
(l)年報要求期間的生產數量
(m)年報要求期間的庫存數量
(n)年報要求期間的銷售美國數量
特別提醒:如上法規規定的激光產品FDA注冊可知,檢測過程描述和檢測結果是注冊最重要的信息之一,也即要提供21CFR 1040.10檢測報告。中為檢驗技術擁有一流激光實驗室,出具檢測報告可以直接用于激光FDA注冊。
產品報告FDA后將的到收信回執(紙質或郵件)。
回執旨在通知報告人:報告已經收到并已錄入數據庫,并分配報告登錄號,年度的注冊號將后三位數字000變為001,每年增加一位數,以此類推。
登錄號是FDA數據庫中報告的唯一標識,有助于特定報告的查取。此外,FDA使用登錄號來確認制造商至少已符合產品的自我聲明符合認證和報告的要求。
登錄號表示FDA已收到報告信,不表示FDA已批準和認可了相關產品,也不表示報告內容的合規和完整新。
特別提醒:FDA注冊執行先分配注冊號,后隨即抽查的監督措施。也即提交虛假資料也能獲得注冊號,但一旦被查收,注冊號即有被取消的風險,企業也將面臨黑名單風險。中為檢驗技術以嚴謹真實的原則,以符合性檢測為前提,為企業提供合規的激光產品FDA注冊。
第四部分:激光FDA注冊號恢復
當激光產品進行FDA注冊時留給FDA的企業聯絡郵箱中收到如下文件時(有可能是企業自己的郵箱,也有可能是當時代理注冊公司的郵箱),說明注冊信息被FDA抽查,并且不合格,注冊號已被暫停。暫停的后果是,該注冊所涉及的所有激光產品將不能再進行出口,已出口的產品可能會被追責,企業有可能已被進入FDA監管黑名單。
一般國內企業都是注冊時候資料通過代理提交虛假資料,甚至不提交任何資料。針對國內普遍情況及FDA要求,我們提供如下解決方案:
序號 | FDA要求 | 需補充資料 | 客戶需提供 | 中為檢驗技術 |
1 | 產品認證符合性證明 | 1.提供質量控制程序文件,包括但不限于產品一致性控制程序、生產流程圖、質檢流程圖、質檢記錄、關鍵元器件控制程序、產品設計圖紙、激光規格書、壽命測試條件和記錄等保持激光產品質量和穩定性的質控文件。 | 1,提供質量控制程序文件,包括但不限于產品一致性控制程序、生產流程圖、質檢流程圖、質檢記錄、關鍵元器件控制程序、產品設計圖紙、激光規格書、壽命測試條件和記錄等保持激光產品質量和穩定性的質控文件。 |
|
2 | 認證標簽 | 1,提供符合21CFR1010.2的認證標簽。或符合Notice 50/56的認證標簽。 |
|
|
3 | 識別標簽 | 1,提供符合要求的產品識別標簽。 | 1,產品識別標簽
| 1,協助完善正確的認證標簽 |
4 | 性能要求 |
| 提供上述1.2.3要求的材料 | 協助整理性能要求的符合性證明說明材料 |
5 | 適用激光類別的警告 |
|
提供以上標簽在產品上標記的照片 | 協助整理并修正標簽,使其符合標準要求 |
5 | 美國代理商 | 提供美國當地代理公司信息及代理授權協議書 | 提供美國當地代理公司信息及代理授權協議書 |
|
7 | 生產情況 | 1,提供所有激光輻射產品的型號、生產數量、庫存數量、銷美數量的統計表 | 提供所有激光輻射產品的型號、生產數量、庫存數量、銷美數量的統計表 | 協助整理符合要求的統計表 |
特別提醒:FDA輻射產品管制是一件嚴肅且有一定繁瑣性的事情。所以在解決注冊號恢復事項中,企業需提供必要的支持。被抽查不合格即說明首次注冊過程中的敷衍做法已經被核查,應不再以隨意處理的態度對待。




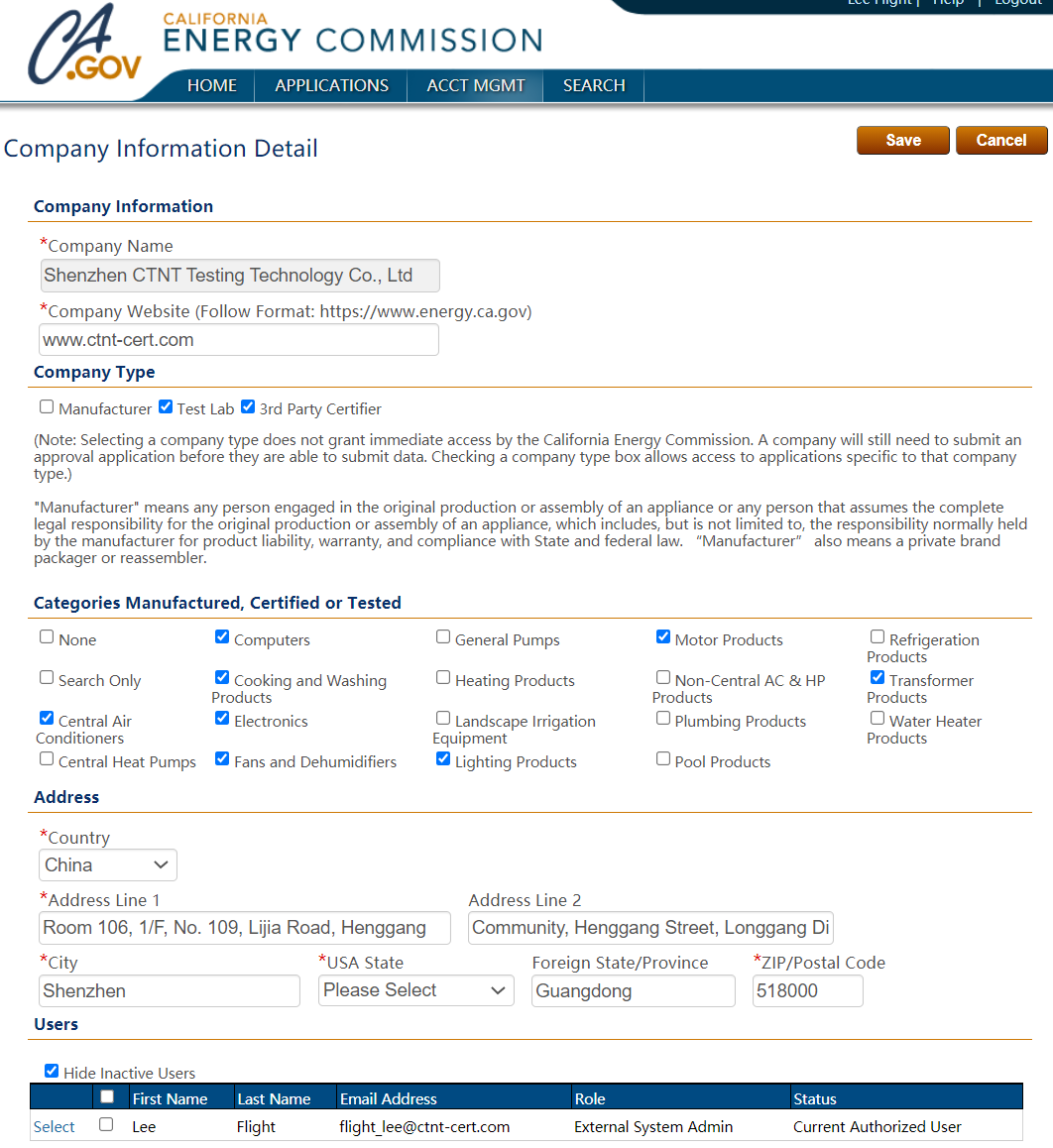
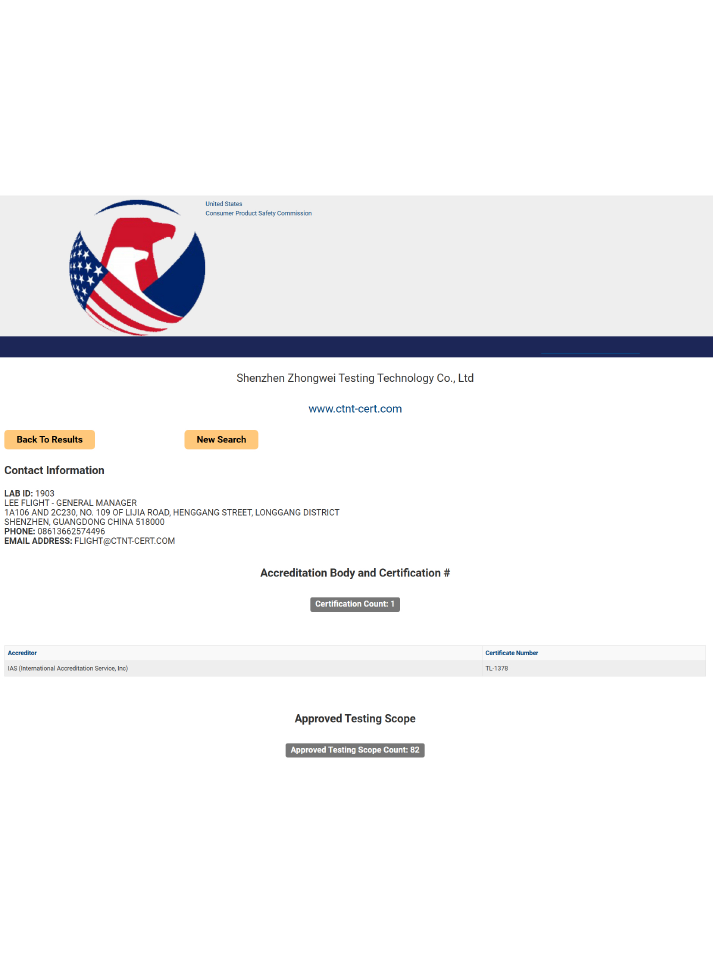

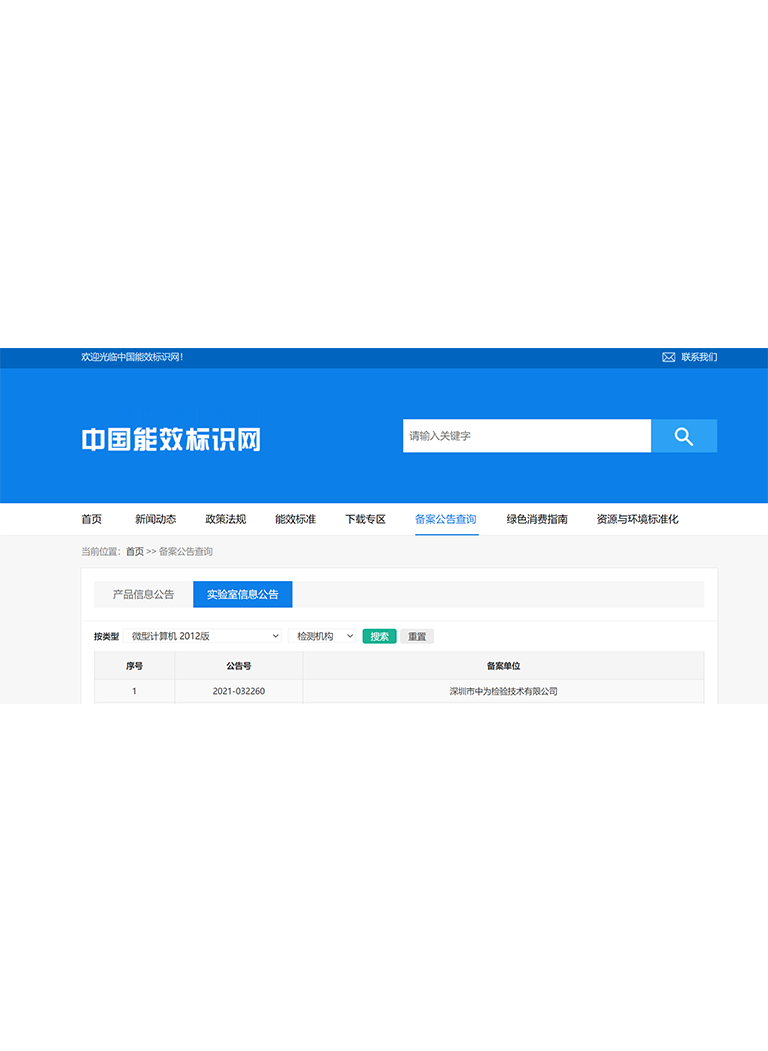
































電話:400-1846-454

地址:深圳市龍崗區橫崗街道橫崗社區力嘉路109號





 網站備案:
網站備案: 公安網備案:
公安網備案:


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}